Cystic fibrosis (CF) is an inherited disease that affects the lungs and the gastrointestinal system but has a broad effect on biochemical mechanisms including amino acid metabolism. Amino acids form the fighters of protein and are essential to muscle growth, immune system and overall health. If you need more interested info like that visit quick guider.
Nonetheless, people affected by CF tend to have amino acid imbalance as a result of malabsorption, chronic inflammation, and altered metabolism. In this article, we discuss the impact that cystic fibrosis has on amino acids, the implications that such interruptions have on the human body, and the possible solutions that one can adopt to ensure proper amino acid levels are maintained.
How Does Cystic Fibrosis Affect Amino Acids?
Cystic fibrosis (CF) is a hereditary disease that covers mostly the lungs and digestive tract, but also other biochemical processes, such as amino acids metabolism. The amino acids are also main components of proteins and are essential in building muscles and immune systems and overall body well being.
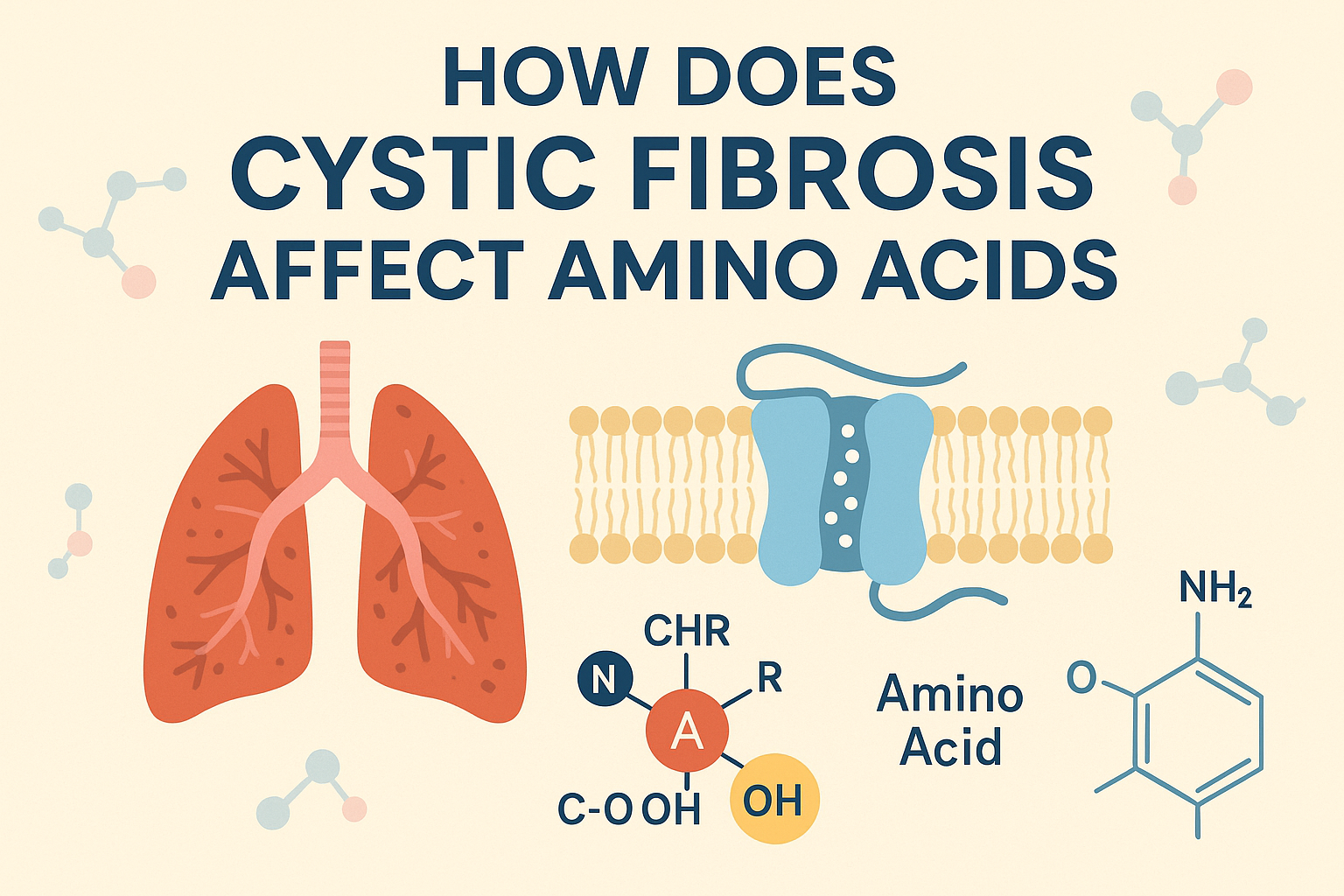
Cystic fibrosis has a direct impact on amino acids since the malfunction of the CFTR protein, an extremely important chloride channel, is known to transport the chloride ions in and out of the cell membrane. Namely, there is a common mutation, termed F508del, which causes a deletion of an amino acid phenylalanine at position 508 in the CFTR protein resulting in its poor functioning and misfolding.
Understanding Cystic Fibrosis and Its Impact on Nutrition
Mutations in the gene, CFTR (Cystic Fibrosis Transmembrane Conductance Regulator) cause cystic fibrosis, which increases the thickness of mucus and causes it to block various organs. The digestive system is one of the most affected systems where the pancreatic system is unable to produce digestive enzymes with the result being malabsorption of nourishment including proteins and amino acids. If you need info related How to reset oil change light.
Patients with CF require additional calories, as much as 200 percent of the levels of those without CF, to combat impaired absorption of nutrients. Enzyme supplements are imperative in digesting food and enhancing the uptake of nutrients.
What Does the CFTR Protein Do?
The CFTR protein acts as a chloride ion channel to take the chloride in and out of the cell, particularly in the lungs where it aids to stabilize the balance between salt and water. Exiting the cell via the CFTR channel draws on the chloride ions, and also draws water, forming a coating in which the cilia on lung cells then run the mucus out of the airways.
In individuals with cystic fibrosis, errors in the CFTR gene cause the protein to become defective so that it cannot transport chloride and can transport water. This causes thick and sticky mucus in the lungs, digestive system and sweat glands causing serious health complications.
How Do Problems With the CFTR Protein Cause CF?
In individuals with cystic fibrosis (CF), CFTR gene mutations may cause an incorrect/incorrectly-functioning, improperly-expressed, or un-expressed protein. This makes the chloride ions to be abnormally trapped within the cells and as a result water fails to moisturize the mucus resulting in the mucus to be thick and sticky. The sticky mucus makes the cilia flat flattened which inhibits ability to remove germs and trash debris, causing them to have difficulty in breathing as well as frequent infections.
Consequently, persons with CF are likely to experience incessant respiratory ailments, poor digestion, and other health problems in other organs.
Cystic Fibrosis and Amino Acids
Individuals with cystic fibrosis (CF) utilize more energy to breathe and defend against infections but cannot break down protein efficiently because of their pancreatic problems, so they lose muscle. Although marginally effective in boosting muscle protein synthesis, adding calorie and protein intake, it has been found that the supplementation of essential amino acids has a higher level of success especially avoiding the processing of digestive enzymes.
There is evidence that free essential amino acids stimulate protein synthesis better than complete protein sources such as whey, particularly in CF. The maintenance of muscle volume is a decisive factor in CF because it enhances lung performance, survival, endurance exercise, and quality of life.
Conclusion
Cystic fibrosis has serious consequences in amino acids because it causes nutrient dietary disorders and more energy requirements in the body, which alters the muscles and the rest of the body health. Malfunctioning CFTR protein not only leads to accumulation of thick mucus in the body organs but also to poor digestion, especially of proteins and amino acids.
Amino acid supplementation with essential amino acids has had potential in enhancing muscle protein synthesis that does not depend on digestive enzymes. The amino acids are vital in maintaining the optimal levels that blend well in managing the symptoms of CF, maintenance of muscle mass and enhancement of quality of life.
FAQS
What amino acid is affected by cystic fibrosis?
The most affected amino acid in cystic fibrosis is phenylalanine. A common mutation called F508del causes this amino acid to be missing from the CFTR protein, making it malfunction.
What amino acids change in cystic fibrosis?
In cystic fibrosis, the mutation often leads to changes in several amino acids, particularly the deletion of phenylalanine at position 508 in the CFTR protein. This affects how the protein folds and works.
How does cystic fibrosis affect protein function?
Cystic fibrosis causes the CFTR protein to malfunction, either by not being made correctly or not working properly. This disrupts the movement of chloride ions across cells, which affects many body functions, especially in the lungs and digestive system.
How does amino acid deletion affect the structure of the CFTR protein?
When phenylalanine is deleted from the CFTR protein, it causes the protein to fold incorrectly. This incorrect folding prevents the protein from functioning as it should, which impacts the transport of chloride ions and leads to the symptoms of cystic fibrosis.